


Cards In This Set
| Front | Back |
|
ICH
|
1990-International Conference on Harmonization for technical requirements of registration of pharmaceuticals for human use (hosted by EFPIA)1989-Initiated at WHO conference International Conference of Drug Regulatory Authorities (ICDRA) as trilateral talks (EU-US-Japan) with IFPMA-Joint RA-Industry initiative for harmonization of standards dealing primarily with Pharmaceutical Regulation
|
|
EFPIA
|
European Federation of Pharma Industries and Associations
|
|
IFPMA
|
International Federation of Pharma Manufactures and Associations
|
|
WHO
|
World Health Organization (Health Care branch of UN)
|
|
ICH E2E
|
2005-PHARMACOVIGILANCE PLANNING. E2E
|
|
CTD/eCTD (ICH M4)
|
Common Technical Document (electronic CTD)-Common dossier structure for pharma submission2004-Current Version2003-Mandatory in EU and Japan, highly recommended in US and Aus
|
|
CTD Organization
|
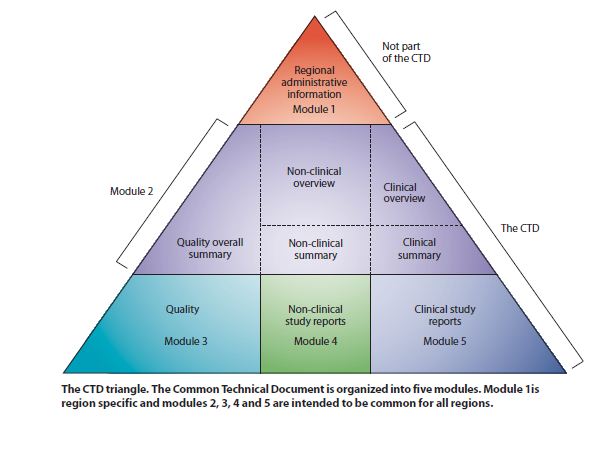 Pyramid structure based on imput from 3 Expert Working Groups (EWG): Modular StructureM1: CV of experts, Risk Management + Regional Requirements especially language/labeling requirementsM2: CTD TOC (2.1); Introduction (2.2), QOS (2.3), NonClinical Overview (2.4) and Summaries (2.6); Clinical Overview (2.5) and Summaries (2.7)M3: Quality (CMC/3.1 TOC) [Not totally harmonized]M4: Non-Clinical Study Reports (4.1 TOC)M5: Clinical Study Reports (5.1 TOC)EWG are:M4Q-Quality (CMC; pharmaceutical data; biologics/biotech)M4E-Efficacy (Clinical; Efficacy)M4S-Safety (Non-clinical Pharmocology and Tox) |
|
ICH Drug Development based on "Core Dossier" Structure
|
Drug Development Components:1. Chemical or Biological Development (CMC)2. Pre-clinical Pharmacology (PK/PD) and Toxicology (GLP-Tox) and POC Efficacy3A. PhI-Safety (15-10 healthy volunteers) looking for PK/PD, dose range finding, tolerability; could include disease patients in some incidences (Ph1/2)3B. Ph2-(50-200 patients) POC/POC target, PK, safety, efficacy (clinical and target through biomarkers); sometimes multiple dose3C. Ph3- (Large-1000's) randomized, typically double-blind study for statistical efficacy and safety (tox) [FDA requires 2 Ph3 studies for NDA)4. Manufacturing Site Approval (GMP compliance)5. Ph4 (may be required for approval) and Post-market surveillance
|
|
ICH Module 3
|
ICH Q8 (R1) Pharmaceutical Development-CMC not totally harmonized -Covers Substance (3.2.S)1. Drug Substance (API)2. Info on manufacturer (name and address), manuf. process and controls, control of materials and critical steps, process validation, manufacturing process development3. Characterization4. Control of drug substance5. Reference Standards6. Container closure system7. Stability-Covers Product (3.2P)1. Description/Comp DP2. Pharma Development (name, formulation, overages and excipients; see ICH Q8) based on sound scientific principles (note, overages to compensate for manuf or shelf life degradation NOT recommended)3. Manufacturer: including who, batch formula, process, control of critica steps/intermediates, process validation4. Control of exciients5. Control of drug product: (specification, analyticals, batch analysis, impurities not completely harmonized and justifications of specifications), KNOW REGIOAL DIFFERENCES
|
|
ICH Module 4
|
ICH S7A GuidanceCovers Preclinical Testing (4.1, 4.2, 4.3)---usually requires at least 2 mammalian species for FDA4.2.1 Pharmacology: PK/PD, safety pharm, PD drug interactions4.2.2 PK: ADME, PK drug interactions, others4.2.3 Tox: Single-dose; repeat-dose tox and genotox (in vitro&in vivo); short-term/long-term carcino; repro/embryo tox; local tolerance; ADA; mechanistic studies; dependence; impurities and metabolites (not completely harmonized)
|
|
ICH Module 5
|
Covers Clinical (5.1-5.4)5.2 Tabular list of all clinical stuides5.3.1 Biopharmaceutical Studies: bioavail(BA) and bioequiv(BE); bioanalytical methods5.3.2 PK using human biomaterials:5.3.3 PK (healthy and patient tolerability; intrinsic factor PK test, extrinsic factor PK test)5.3.4 PD (healthy and patient PK/PD studies)5.3.5 Efficacy and Safety (controlled and uncontrolled clinical studies; analysis against claims and endpoints)5.3.6 Postmarket experience: Periodic Safety Update Reports for approved products; PV results5.3.7 Case Report Forms and Individual Patient Listing
|
|
MAA
|
Marketing Authorization Application (FDA NDA)-Full standalone application; complete application-Must include physiochemical, biological or microbiol tests; PK/PD and Tox; Clinical Trials-Usually required for NCE/NME (chemical/biologic/radiochem not previously authorized) as well as others, including new fixed combinations
|
|
MAA Mixed Data
|
Somewhat abbreviated application for certain products with significant published literature-Still considered full and complete
|
|
MAA Bibliographic Reference
|
Re-introduction of old NCE (on the market for at least 10 years)-CMC required-PK/PD and clinical can be referenced Does not apply to new indication
|
|
MAA Abridged
|
Generic NCE (not universal for biologic/biosimilars yet)Must be essentially equivalent (API is the same; same pharmaceutical form; Bioequivalent or Therapeutic Equivalence (FDA)-M1, M2, and M3- original dossier must be available to reviewing CA/RA- IF Not Off-Patent, Innovator grants rights to information in original dossier- Written comfirmation from innovator is required, granting generic rights
|
